(4598)Delta-Fly Pharma株式会社 減益、契約一時金等の収益に期待
|
 江島 淸 社長 |
Delta-Fly Pharma株式会社(4598) |
|
 |
企業情報
|
市場 |
東証マザーズ |
|
業種 |
医薬品(製造業) |
|
代表取締役社長 |
江島 淸 |
|
所在地 |
徳島県徳島市川内町宮島錦野37-5 |
|
決算月 |
3月末日 |
|
HP |
株式情報
|
株価 |
発行済株式数 |
時価総額 |
ROE(実) |
売買単位 |
|
|
1,548円 |
5,419,600株 |
8,389百万円 |
-41.7% |
100株 |
|
|
DPS(予) |
配当利回り(予) |
EPS(予) |
PER(予) |
BPS(実) |
PBR(実) |
|
0.00円 |
– |
-239.87円 |
– |
390.87円 |
4.0倍 |
*株価は12/9終値。発行済株式数、DPS、EPSは22年3月期第2四半期決算短信より。ROE、BPSは前期実績。
業績推移
|
決算期 |
売上高 |
営業利益 |
経常利益 |
当期純利益 |
EPS |
DPS |
|
2018年3月(実) |
150 |
-243 |
-244 |
-246 |
-71.20 |
0.00 |
|
2019年3月(実) |
– |
-592 |
-671 |
-673 |
-170.16 |
0.00 |
|
2020年3月(実) |
100 |
-1,545 |
-1,552 |
-1,555 |
-348.32 |
0.00 |
|
2021年3月(実) |
300 |
-852 |
-859 |
-862 |
-187.34 |
0.00 |
|
2022年3月(予) |
100 |
-1,300 |
-1,300 |
-1,300 |
-239.87 |
0.00 |
*単位:百万円、円。予想は会社側予想。2018年6月25日付で1:500の株式分割を実施。EPSは遡及調整。
Delta-Fly Pharma株式会社の業績動向、開発状況の進捗などをご紹介します。
目次
今回のポイント
1.会社概要
2.業績動向
3.成長戦略
4.今後の注目点
<参考:コーポレート・ガバナンスについて>
今回のポイント
- 既存の抗がん活性物質等を「モジュール」(構成単位)として利用し、用法用量や結合様式等に創意工夫を加えて組み立てることで臨床上の有効性と安全性のバランスを向上させた副作用の少ない新規抗がん剤を創製する「モジュール創薬」という独自コンセプトで抗がん剤を開発。
- 「モジュール創薬」は治療効果の向上、副作用軽減、低コストといった患者メリットに加え、特許化による高い排他性、迅速な開発スピード、低開発リスクといった開発上のメリットも大きい。同社では現在6つの製品・開発パイプラインを有し、4品目が臨床試験実施中、2品目が臨床試験準備中である。
- モジュール創薬の他、抗がん剤開発への特化、経験豊富なメンバーによる開発、外部資源の有効活用による効率的な企業運営なども同社の特徴。
- 22年3月期第2四半期の事業収益は100百万円。日本ケミファとのライセンス契約によるマイルストーン収入を取得した。事業費用は、前年同期比16.1%増の6億54百万円。開発パイプラインの臨床試験における医療機関並びに症例数の増加、次試験に向けた治験薬となる原薬や製剤の製造などを進めた。営業損失は前年同期比90百万円拡大の5億54百万円となった。
- 22年3月期の事業収益は、ライセンス契約に伴うマイルストーン対価として前期比2億円減少の1億円を見込んでいる。DFP-10917のマイルストーン対価による収益のほか、米国で臨床第III相試験を実施しているDFP-10917や、国内で臨床第II相試験を実施しているDFP-14323を含め、複数の抗がん剤候補化合物の臨床試験が進んでおり、新しいパートナーとの提携による契約一時金等の収益も期待している。今後、収益が確実になった段階で適時見通しを明らかにしていく予定。事業費用は前期比2億50百万円増加の14億円の計画。各種開発パイプラインを着実に進めるため、研究開発費は同2億23百万円増加する見込み。営業損失は前期比4億47百万円拡大の13億円の予想。
- 引き続き、創業11年目に入った同社の具体的な成果として、最短での申請が予定されているDFP-10917の「2023年3月期上期の申請、同期下期の上市」に向けた着実な進捗を期待したい。
1.会社概要
『「がん」だけを見ることなく、「がん患者」の全体を診ることにより、安心して家族のがん患者に勧められる治療法を提供すること』を企業理念に、既存の抗がん活性物質等を「モジュール」(構成単位)として利用し、用法用量や結合様式等に創意工夫を加えて組み立てることで臨床上の有効性と安全性のバランスを向上させた副作用の少ない新規抗がん剤を創製する「モジュール創薬」という独自コンセプトで抗がん剤を開発。
【1-1 沿革】
徳島県出身の江島社長は、名古屋工業大学卒業、東京工業大学修士課程修了後、地元徳島県の製薬企業である大塚グループに就職し、その事業会社の一つ大鵬薬品工業に配属となった。入社後すぐに早稲田大学理工学部に留学し、約12年間、研究者として医薬品、特に、機能性高分子から成る新薬の開発に関する研究に取り組む。その後、大鵬薬品工業の医薬品のシーズ探索を担当する部門在籍時、米国バイオベンチャーのマネジメントのあり方などを目の当たりにした際、大手製薬企業の研究開発組織で開発に携わるのではなく、独立して自分の力で製薬会社をマネジメントし、新しいアプローチで創薬を行いたいという意欲が強く湧き上がる。同時に、単に創薬を目指すのではなく、目の前にいる患者に何をしてあげられるのかを常に考えながら、ビジネスとして成立させることを目指し、2010年、江島社長61歳の時、大鵬薬品工業を退任し、Delta-Fly Pharma株式会社を設立。モジュール創薬による副作用の少ない患者に優しい抗がん剤開発に取り組んでおり、2020年9月現在6つの製品・開発パイプラインを有している。2018年10月、東証マザーズに上場した。
【1-2 企業理念・経営理念】
社名「Delta-Fly」は「Dragonfly(とんぼ)」に由来している。とんぼは前にしか進まず退かないところから「不退転」の精神を象徴し、「勝ち虫」とも呼ばれていることから、強い意志をもって医薬品開発を行う決意を表している。
|
企業理念 |
「がん」だけを見ることなく、「がん患者」の全体を診ることにより、安心して家族のがん患者に勧められる治療法を提供すること |
後述するように、同社は「がん」に打ち勝つことのみを目的とする抗がん剤を開発するのではなく、抗がん剤の大きな課題である副作用を軽減し、価格も含め患者およびその家族が安心して用いることのできる抗がん治療を提供することを自社の社会的存在意義であると認識している。
【1-3 同社を取り巻く環境】
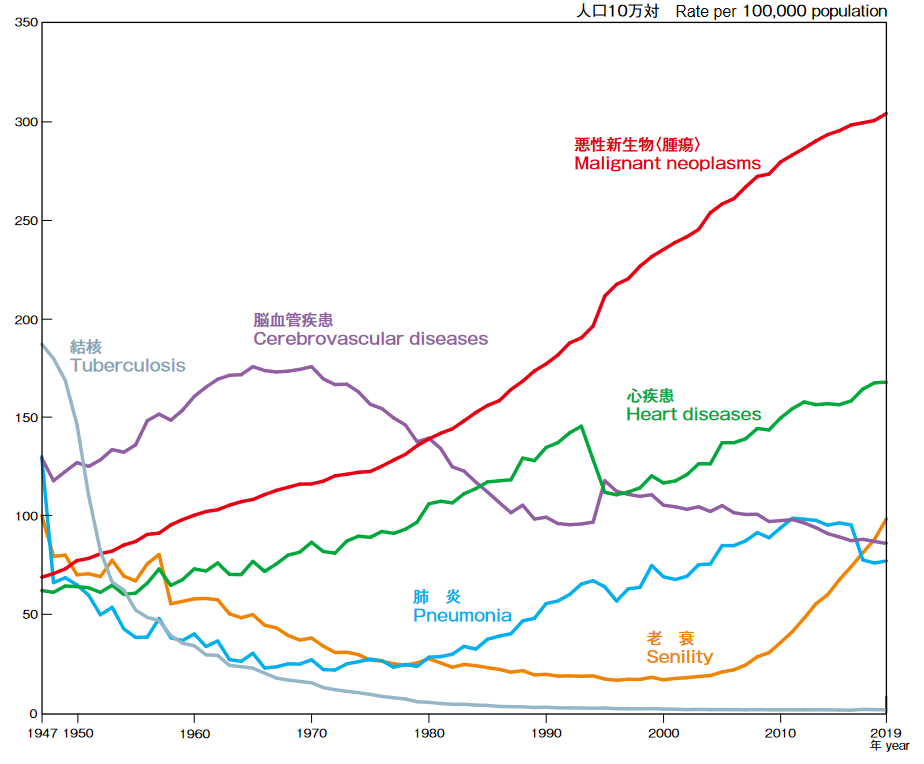
(1)増加傾向が続くがん死亡者数
公益財団法人がん研究振興財団「がんの統計2021」によれば、がん(悪性新生物)は1981年から死因の第1位を占め、2019年の死亡者は37万6,425人、人口10万対死亡率304.2であり、総死亡の27.3%を占めている。高齢化、また食生活を含めたライフスタイルの変化等によりがん発症率は上昇していると言われている。
(同社資料より)
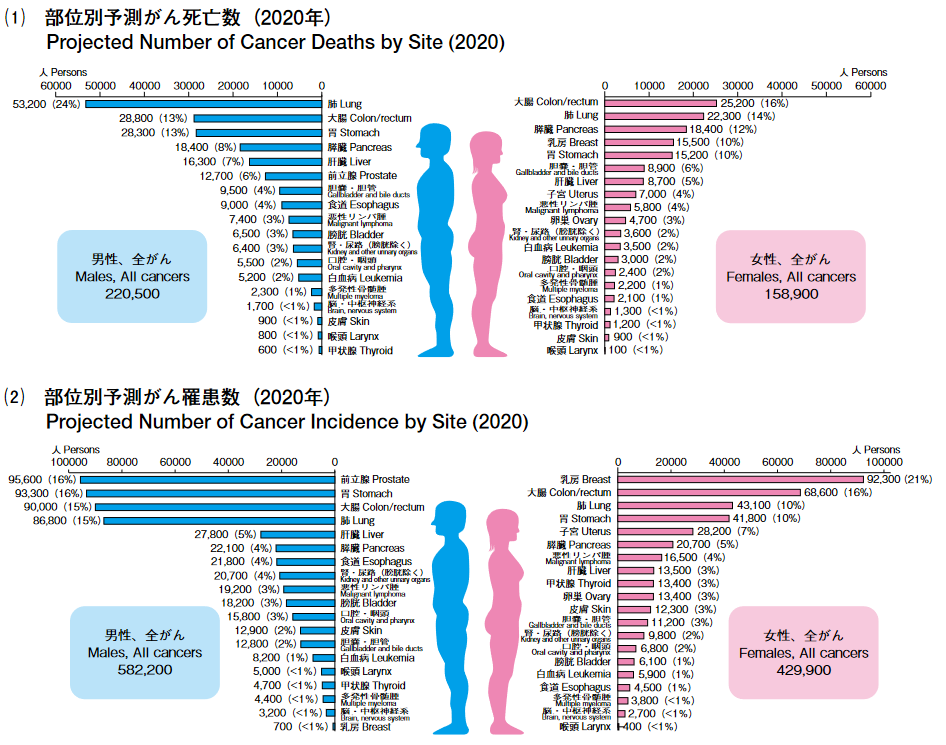
わが国のがん死亡数の2020 年推計値は、約37万9400人である(男性22万500人、女性15万8,900人)。部位別の死亡数は、男性では肺が最も多くがん死亡全体の 24% を占め、次いで大腸(13%)、胃(13%)、膵臓(8%)、肝臓(7%)の順、女性では大腸が最も多く(16%)、次いで、肺(14%)、膵臓(12%)、乳房(10%)、胃(10%)の順となっている。
わが国のがん罹患数の2020年推計値は、約101万2,000例である(男性58万2,200例、女性42万 9,900例)。部位別では男性で前立腺(16%)、胃(16%)、大腸(15%)、肺(15%)、肝臓(5%)の順、女性で乳房(21%)、大腸(16%)、肺(10%)、胃(10%)、子宮(7%)の順となっている。
(同社資料より)
(2)がんの治療方法
「がん」の主な治療方法には「手術療法」「放射線療法」「化学療法(抗がん剤治療)」があり、抗がん剤投与の「化学療法」は手術や放射線で治療後の第3の治療。
「がん」の進行度ステージ3~4の患者に対しては一般的に標準療法(大規模な臨床試験によって、治療効果の可能性が示され、かつ安全性が許容された、最も推奨される治療法)である抗がん剤投与が行われる。
(3)抗がん剤開発と副作用
世界的ながん患者の増加に対し欧米、日本の各企業が様々な抗がん剤開発を進めているが、周知のように抗がん剤治療に伴う各種副作用は、がん患者にとって大きな負担であり、患者のQOL(Quality Of Life:生活の質)向上の観点から副作用の軽減ニーズは極めて大きなものとなっている。
(副作用発生の仕組み)
がん細胞は、急速に分裂して成長するので、抗がん剤は、成長の速い細胞を殺すように作られている。しかし同時に健康な細胞にも、骨髄で造られる血液細胞、消化器の細胞、生殖器の細胞、毛根細胞など急速に細胞分裂するものがあり、抗がん剤はがん細胞だけでなくこれらの正常細胞にも影響を与えてしまい、嘔気、嘔吐、脱毛、疲労感といった副作用を引き起こす。
【1-4 事業内容】
(1)同社の創薬方法:モジュール創薬
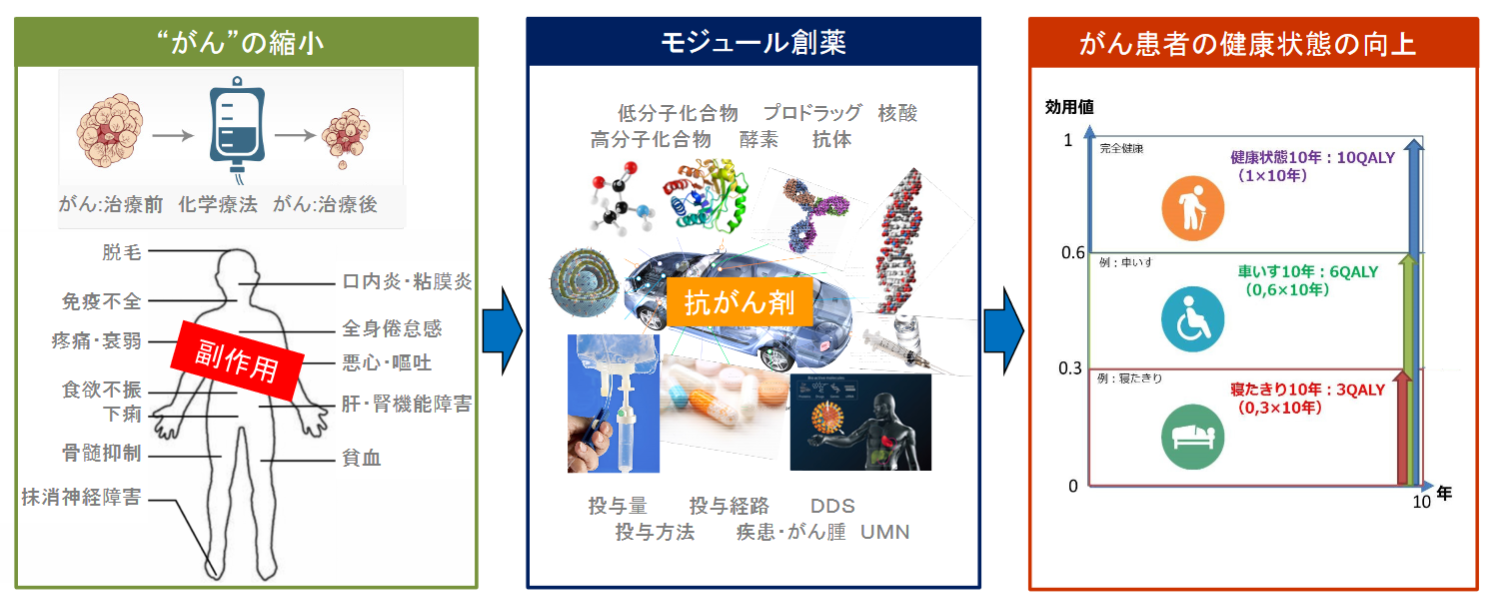
多くのバイオベンチャーがある中で、同社を最も特徴づけるのが同社の創薬コンセプト「モジュール創薬」である。
(同社資料より)
既存の抗がん活性物質等を「モジュール」(構成単位)として利用し、創意工夫(用法用量・結合様式等)を加えて「アセンブリ」(組み立て)することで、臨床上の有効性と安全性のバランスを向上させた新規抗がん剤を創製する手法が「モジュール創薬」である。
「モジュール創薬」では「がん」だけを見ることなく、「がん患者」の全体を診ることによって、未だに効果が限定的で多くの様々な副作用のある抗がん剤を複合的に改良して、副作用の少ない安心して家族のがん患者に勧められる薬剤にする。
(モジュール創薬の優位性)
|
患者へのメリット |
◇ 患者情報に基づく創薬だから治療効果が上がる。 ◇ 患者情報に基づく創薬だから従来の副作用が軽減する。 ◇ 基礎と臨床試験が少なく短期間だからコストが低い。 |
|
開発上のメリット |
◇ 新規性・進歩性により特許化できるから高い排他性を有する。 ◇ 患者情報に基づく開発だから開発スピードが速い。 ◇ 患者情報に基づく開発だから開発リスクが低い。 |
通常一つの医薬品を開発するのには、基礎の探索研究段階でがんの特異的な部分に作用する化合物をスクリーニングし、可能性のある化合物を抗がん剤候補とするが、臨床段階で作用を確認し、臨床試験で有効性と安全性を実証する必要があり、基礎段階からの研究開発に10~15年に亘る長い期間を要する。またそれに伴い、数十億~数百億円にのぼる大規模な資金が必要である。
また、承認に至るまでの各段階において様々な要因により開発中止に至るリスクが大きく、世界の製薬会社や創薬ベンチャー企業にとっては研究開発プロセスの効率化と開発リスクの低減が大きな課題となっている。
これに対して、「モジュール創薬」は、既に医薬品として使用されている抗がん剤の活性物質を利用して組み合わせるため、基礎の探索研究がほとんど不要であることに加え、臨床での有効性と安全性の予測が可能であるため創薬に着手して1~2年後には臨床試験を開始できているなど、一般的な抗がん剤よりも研究開発の効率が高く、開発期間も短くなり、臨床試験で失敗する等の開発リスクが低減されている。
また、がん患者の治療上の課題に注目して、特許切れの医薬品を抗がん剤の知識とノウハウを駆使して組み合わせれば、新規の抗がん剤としての特許化が可能である。

(同社資料より)
また、近年、新薬開発のコスト低減などを目的とし、製薬企業においては後発薬ジェネリックや既存薬剤から新たな薬効を見つけ出すドラッグ・リポジショニングの開発が拡大している。
これらは既存薬を利用するという点では「モジュール創薬」と同じである。ジェネリックはもちろんだが、ドラッグ・リポジショニングにおいても新規性・進歩性が認められにくいため特許取得が困難であるのに対し、「モジュール創薬」は全て特許化された新たな薬剤に生まれ変わるという点が決定的な違いとなっている。
このように、抗がん剤の問題点を解決しようとする限り、完全に新規の抗がん剤を生み出すことが可能であることから、同社では「モジュール創薬」は新たな創薬手法の大きなイノベーションになり得ると確信している。
(2)ビジネスモデル・収益モデル
(ビジネスモデル:効率的な研究開発体制を構築)
新しい医薬品が上市されるまでには、「基礎研究」から始まり、「前臨床試験(動物を用いて薬効薬理作用、生体内での動態、有害な作用などを調べる試験)」、「臨床試験(医薬品や治療技術などの人間への影響を調べる科学的試験)」を経て、当局への申請・承認を得たのち、「製造」、「販売・マーケティング・製造販売後調査」といったプロセスを経るのが一般的である。
こうしたプロセスにおいて同社は、研究開発のマネジメント業務に集中し、具体的な業務については国内外の優れた外部の研究開発受託会社や製造受託会社に委託しており、開発フェーズに応じた外部協力機関との連携により、効率的な研究開発体制を実現している。
また、三洋化成工業株式会社(東証1部、4471)との間で、ドラッグデリバリーシステムを用いた新規抗がん剤における共同研究開発にも取り組んでいる。
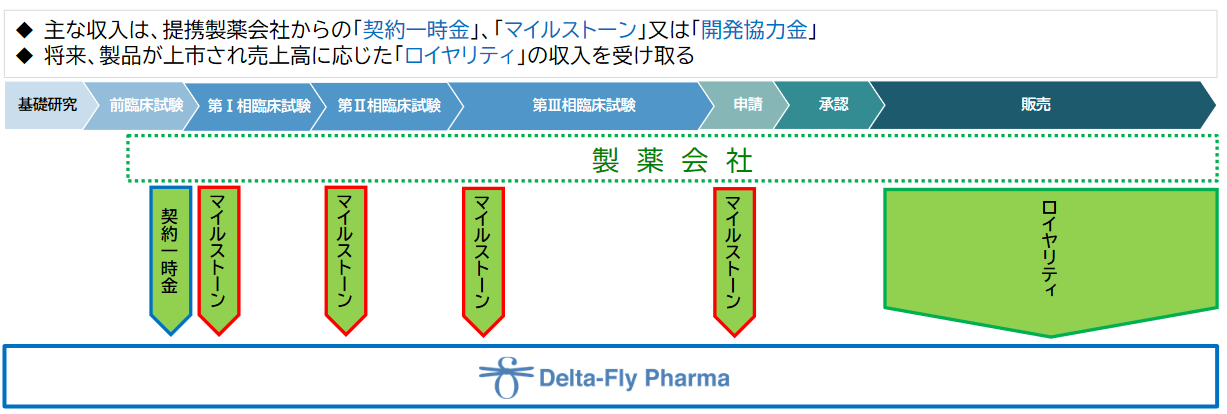
(収益モデル)
研究開発段階においては、提携製薬会社との契約に基づく「契約一時金」、「マイルストーン」、「開発協力金」が主な収入となる。将来、提携対象の製品が上市に至った場合には、売上高に応じた「ロイヤリティ」収入を受け取る予定である。
|
収益名 |
内容 |
|
契約一時金 |
契約一時金として受取る収入 |
|
マイルストーン |
研究開発の進捗に応じて、事前に設定したイベントを達成した際に受取る収入 |
|
開発協力金 |
研究開発費用に応じ、提携会社が負担する分の収入 |
|
ロイヤリティ |
医薬品販売後に売上高に応じて受取る収入 |
現在のライセンス導出契約は以下の通り。
|
相手先 |
契約内容 |
契約期間 |
|
日本新薬株式会社 (東証1部、4516) |
DFP-10917の 独占的特許実施許諾 |
日本における特許権が消滅するまで又は販売開始後15年のいずれか遅い方まで |
|
日本ケミファ株式会社 (東証1部、4539) |
DFP-17729の 独占的特許実施許諾 |
日本ケミファ(株)及びサブライセンシーが本製品の販売を終了するまで |
(同社資料より)
(3)製品・開発パイプライン
現在、前述の経営方針に沿って以下6つの製品・開発パイプラインを有している。
パイプラインの開発・事業化の経緯、現状、今後の計画は以下のとおりで、4品目が臨床試験実施中、2品目が臨床試験準備中である。

(同社資料より)
①「DFP-10917」
|
項目 |
概要 |
|
主な対象疾病 |
難治性・再発急性骨髄性白血病 急性骨髄性白血病(AML)の年間死亡者数は日本1万人、米国3万人、欧州3万人、中国2万人。 白血病による死亡者の85%は60歳以上である。 標準療法は確立されており、一時的には7割程度は血液中のがん細胞が消滅する寛解となるが、再発も多く、完全に治癒するのは全体の3割である。 |
|
既存薬の特徴など |
既存薬CNDACは固形がんを対象疾患とし、投与量は高用量・短時間で、投与経路は点滴または経口。固形がんへの効果が限定的であるのに加え重篤な副作用が散見された。 |
|
モジュールの改良点・効果 |
投与量を低用量・長時間とし、投与経路も点滴による持続静注としたところ、従来使用されてきている核酸誘導体(シタラビンやゲムシタビンなど)とは異なる作用を引き起こし、既存の化学療法が無効な難治性・再発急性骨髄性白血病患者に対しても、薬効を期待できる。 効果と安全性のバランスに優れ、末期の血液がんの治療に最適である。 |
|
特許取得国(21年5月) |
日本、アメリカ、EU、中国、オーストラリア、韓国、ロシア |
(開発状況・今後の事業化)
米国で行われた臨床第I/II相試験では、第II相パートで48%(14/29例)の患者で完全寛解し、高い有用性が示唆された。
これを受け、米国規制当局(FDA)との臨床第II相試験終了時会議を経て、臨床第III相試験の治験実施計画書を提出。合意を得ることができたが、再発・難治急性骨髄性白血病の治療体系の変更に伴い第III相試験のプロトコールの一部を改訂の上、米国FDAに再提出し、臨床第III相試験が始まった。スタートアップミーティングを実施し、被験者スクリーニングを開始した。
新型コロナウイルス感染症の影響も考慮し、症例登録を推進するため治験参加病院数を39病院に増やし、臨床第III相試験を進めている。DFP-10917の新薬承認申請用の原薬と最終製剤は既に確保済みであり、「2023年3月期上期の申請、同期下期の上市」を目指している。
21年5月には、新薬承認取得に向けた準備の一環として、世界保健機関(WHO)の医薬品国際一般名称委員会より、「DFP-10917」の国際一般名称が「Radgocitabine(ラドゴシタビン)」と決定された。
医薬品国際一般名称の決定は、新薬承認取得のための重要な手続きであり、新薬承認後は世界共通の固有名称として使用される。
日本国内については、ライセンス先の日本新薬株式会社が医薬品医療機器総合機構(PMDA)へ2021年1月8日に臨床第1相試験開始のために治験届けをしていたが、2月8日をもって、PMDAから 難治性又は再発のAML患者を対象に日本国内での臨床第1相試験実施の許可を得た。
日本以外のテリトリーでの販売権に関しては、欧米の製薬会社及び中国の製薬会社とライセンス契約交渉中。
(特許関係)
DFP-10917「ラドゴシタビン」とVenetoclax(VTX、ベネトクラクス)の新規誘導体の併用療法及び併用剤に関する発明を世界の主要国に特許出願中だが、2021年6月、日本において特許査定された。
VTXの新規誘導体は、Venetoclaxを水溶性の高分子に共有結合させた新規物質で、標的部位のがん病巣に活性物質のVenetoclaxを選択的に輸送できるため、ヒト急性骨髄性白血病細胞を皮下移植した動物実験では、既存のVTXの投与量の数十分の一以下で同等の薬効を示し、安全性に優れていることを確認した。
2023年3月期中に予定しているDFP-10917「ラドゴシタビン」単剤の米国における製造・販売承認後、世界の主要国における市場の最大化と独占的販売期間を延長する目的で、DFP-10917「ラドゴシタビン」とVTXとの併用療法の臨床試験に実施する予定である。
②「DFP-17729」
|
項目 |
概要 |
|
主な対象疾病 |
末期の膵臓がん、悪性黒色腫胃リンパ腫、胃がん、肺がん すい臓がんの年間死亡者数は、世界で約47万人、日本で約3.7万人。 |
|
既存薬の特徴など |
既存薬である尿アルカリ化剤は、高尿酸血症などを対象疾患とするものだが、膵がんで延命効果が認められたほか、各がん腫瘍で抗腫瘍効果が見られた。 |
|
モジュールの改良点・効果 |
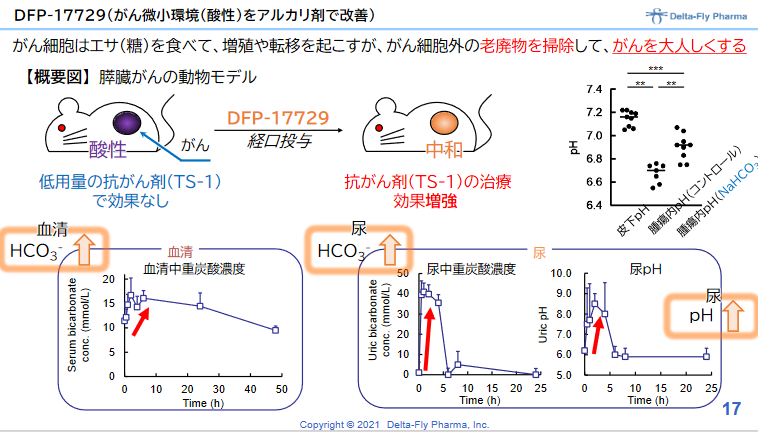
正常細胞では細胞内と比べて細胞外でアルカリ性となっているが、がん細胞の細胞外は酸性となっている。これは、がん細胞の増殖により解糖系が亢進し、乳酸や水素イオンが産生され、それを積極的に細胞外へ排出しているからである。 DFP-17729は、がん細胞の細胞外をアルカリ化することにより、がんの増殖を抑える。いわば、がんの周りを掃除し、がんを大人しくさせるものである。 抗がん剤との併用、免疫チェックポイント阻害剤との併用により免疫チェックポイント阻害薬単独療法に比べて効果を増強することが動物実験で確認されている。 |
|
特許取得国(21年5月末) |
日本、韓国、中華民国 |
◎DFP-17729の臨床効果の確認
|
 |
 |
(同社資料より)
(開発状況・今後の事業化)
医薬品として承認・販売されている尿アルカリ化剤の、日本における抗がん剤としての適応追加の準備を進めている。
尿アルカリ化剤は「アシドーシスの改善」の効能・効果で、「高尿酸血症」や「腫瘍崩壊症候群」などの治療で、すでに臨床現場で使用されているため、前臨床試験は不要。抗がん剤や免疫チェックポイント阻害剤との併用により、既存薬の抗腫瘍効果の範囲を広げ、新たながん治療の提供を目指す。
2020年3月、日本国内における「DFP-17729」の独占的販売権ならびに日本国内で販売するための独占的製造権を日本ケミファに付与するライセンス契約を締結することを合意した。
Delta Fly Pharmaは既存の抗がん剤との併用で膵臓がん患者を対象に臨床試験を実施し、日本ケミファは日本において「DFP-17729」の製造承認が取得された後の販売と製造を担う。
2020年5月、「DFP-17729」に関する論文を米国がん学会誌「Molecular Cancer Therapeutics」に投稿した。
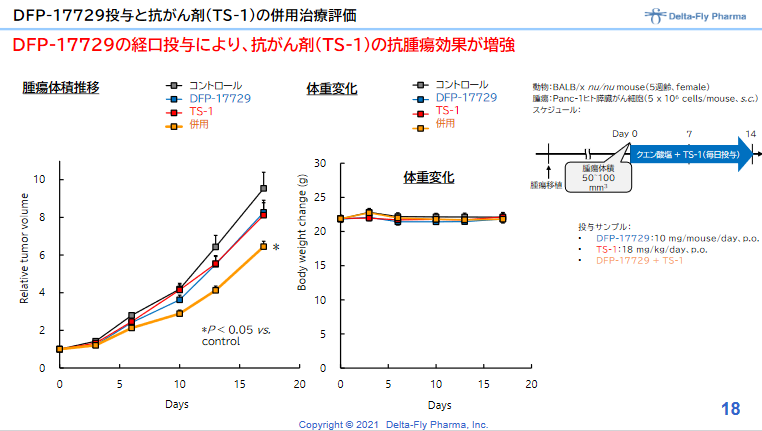
一般に膵臓がん患者の5年生存率は数%以下と悲惨な状況にあるが、この研究では既存の膵臓がん治療剤の治療効果を高めるとともに、がん免疫チェックポイン阻害剤(抗PD-1抗体)の効果を高めることが示されている。また、「DFP-17729」は、既存の抗がん剤に見られる副作用はなく、既存の抗がん剤との併用による毒性の上乗せを伴わないことも確認している。
こうした実績を受け、末期のすい臓がん患者を対象に日本国内の複数の医療機関において、臨床第1相/第Ⅱ相試験を実施することを目的に、2020年7月には治験計画届をPMDA(独立行政法人医薬品医療機器総合機構)に提出し、PMDAの調査も完了し実施が許可された。
この臨床試験は、末期の膵臓がん患者の病状を鑑み、臨床第3相試験に移行する前に臨床第1相/第2相試験での安全性/有効性を探索的に確認するもので、第1相部分では既存薬とDFP-17729を併用した場合の安全性を確認し、第2相部分では既存薬と比べて優れているかを確認する比較試験を行うもの。
2020年11月18日に関東地区の大手病院(3病院)において臨床第1相試験部分の症例登録が開始され、同試験を進めてきたが、第1相試験部分の登録症例全例の安全性評価期間が終了し、2021年4月15日に開催された安全性評価委員会による審議の結果、本剤と抗がん剤併用時の安全性が確認され、第2相試験部分への移行が決まった。
大手病院(6病院)で開始された臨床第2相試験では、同年4月22日に第1症例への投薬が開始された。
この試験は臨床第1/2相試験として実施しているため、安全性評価委員会における第1相試験部分の安全性の確認から第2相試験部分の投薬開始までほぼ間隔を置かずに実施に至ることができた。
その後、2021年11月11日付で症例登録が完了した。今後半年間で、DFP-17729 の有用性を検証し、その結果次第では、独立行政法人医薬品医療機器総合機構(PMDA)へ承認申請を行うことが可能か、あるいは臨床第3 相試験の準備に取り掛かるかの判断を行う。
一方、2021年1月には、ヒトの膵臓がん細胞を移植した動物を用いて、がん微小環境改善剤である「DFP-17729」によって膵臓がん治療薬のティーエスワンの治療効果が高まることが確認されたと発表した。これにより膵臓がんの適応だけでなく、悪性メラノーマ、胃がん、非小細胞肺がん等の適応取得のための日本における開発技術的基盤と知財基盤が整ったと同社では考えている。
提携パートナーの日本ケミファ(株)と共同で日本における臨床試験を進めた後、将来的には、日本国内の治験データに基づいて、米欧やアジア諸国でも展開する計画である。
現時点では、「2023年3月期中の臨床第3相試験への移行、2025年3月期上期申請、同期下期上市」の予定である。
③「DFP-14323」
|
項目 |
概要 |
|
主な対象疾病 |
末期の肺がんなど。 肺がんの年間死亡者数は世界で約180万人。日本で約7.5万人。 |
|
既存薬の特徴など |
既存薬ウベニメクス(UBX)は血液がんを対象疾患とし、投与量は高用量で、投与方法は単剤。経路は点滴または経口。血液がんのみの適応だが、肺がんで延命効果があった。 |
|
モジュールの改良点・効果 |
抗腫瘍効果の増強を目的とし投与量を低用量、投与方法を分子標的治療薬との併用としたところ、肺がんでの効果が確認された。がん患者の免疫機能を改善し、既存薬を効き易くする。末期又は高齢の固形がん患者の治療が期待できる。 |
|
特許取得国(21年5月) |
日本、アメリカ、EU、オーストラリア、韓国、ロシア、中華民国 |
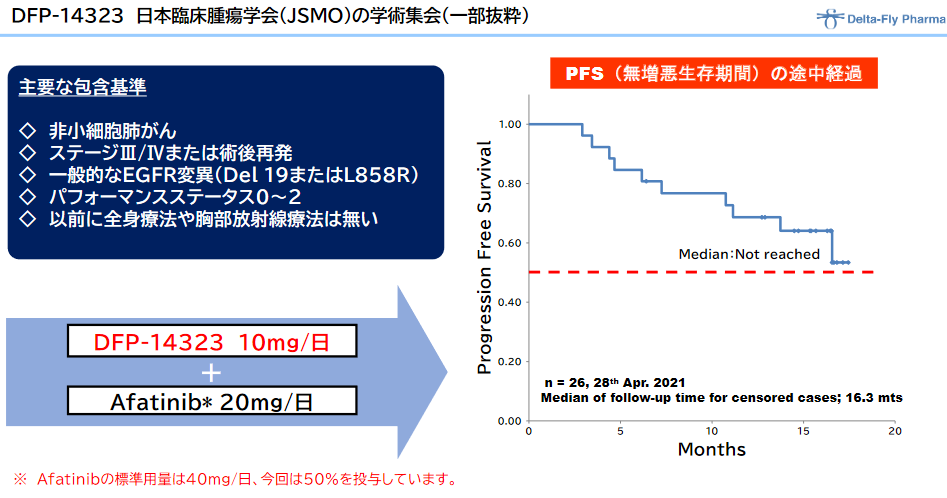
◎「DFP-14323」の臨床効果
(同社資料より)
(開発状況・今後の事業化)
既存薬ウベニメクスは日本において、日本化薬(株)が、「成人急性非リンパ性白血病に対する完全寛解導入後の維持強化化学療法剤との併用による生存期間の延長」の効能・効果で承認済。
Delta-Flyは適応追加として、「EGFR 遺伝子変異陽性非小細胞肺がん患者を対象とした低用量EGFR-TKI 併用治療の臨床第II相試験」を2018年1月から日本国内で開始し、国内治験参加施設の拡大により、新規症例の登録を進めてきたが、2020年3月、症例数全ての症例登録が完了した。
DFP-14323は、ウベニメクスの「新規効能拡大」と「新薬」として承認取得するためのDelta-Flyの開発コード。
その後、登録した全症例(脳転移症例を含む)の病勢コントロール率に基づく効果判定作業を進めていたところ、同年6月には病勢コントロール率100%に達し、また、独立の立場の医師による効果判定評価において、病勢コントロール率(DCR)が100%および奏効率(ORR)が65.4%以上と有効性が確定した。非小細胞肺がんの患者の脳転移に対して優れた治療効果を示したものと同社では考えている。
また、「脳転移を有する末期非小細胞肺がん患者を治療するための組み合わせ医薬品」としても有用であることを改めて見出したことに基づき、特許をPCT(特許協力条約)加盟国に対し国際出願した。
加えて、日本国内における臨床第Ⅱ相試験の結果について、2020年11月に開催されたESMO ASIA CONGRESS 2020(欧州臨 床腫瘍学会アジア大会)のポスターセッションで発表され、臨床第2相試験での高い安全性とPFS(無増悪生存期間)の最新データが示された。
PFS(無増悪生存期間)のデータは臨床第3相比較試験のプロトコールを確定するうえで重要な情報であり、臨床第2相試験の臨床試験データに高い関心を寄せている国内外の製薬会社の協力を得て、2023年3月期開始予定の臨床第3相試験を加速させる予定である。
日本における臨床第2相試験の良好な成績と知財基盤を下に、今後のDFP-14323の臨床第3相比較試験の対象を「脳転移を伴う非小細胞肺がんの患者」に予定し、肺がん患者の数が世界で最も多いとされる中国を含めることで、一日でも早い承認・上市を目指し準備を進めていく。
現時点では「2023年3月期の臨床第3相試験開始、2025年3月期下期の申請、2026年3月期上期の上市」を予定している。
協和化学工業株式会社(未上場)と日本における独占的ライセンス契約を締結していたが、2020年11月、協和化学工業の社内事情により共同開発を断念し契約解除に合意した。
今後はPMDAへのウベニメクスの後発品の製造販売承認を単独で継続するとともに、更に、PFS(無増悪生存期間)とOS(全生存期間)の判定の予定時期(2021年6月頃)を待って、PMDAへのウベニメクスの「効能拡大品」の製造販売承認申請を行う。
(特許関係)
2020年5月、欧州における特許が成立した。
現在、中華人民共和国においてもDFP-14323の特許申請を行っており、中国特許庁との間で審査対応中。中華人民共和国での特許が成立した際には、主要国におけるグローバル事業展開の体制が整う予定である。
④「DFP-11207」
|
項目 |
概要 |
|
主な対象疾病 |
進行再発膵臓がん胃がん すい臓がんの年間死亡者数は、世界で約47万人、日本で約3.7万人。 |
|
既存薬の特徴など |
既存薬ティーエスワンは血小板減少を含む血液毒性により治療の継続が不充分であった。 |
|
モジュールの改良点・効果 |
DFP-11207は抗がん作用を有する5-フルオロウラシル(5-FU)の薬物動態を制御するために、徐放・阻害・失活させる3つのモジュール化された活性物質(モジュールI、II、III)を結合した化合物。 従来の5-FU系抗がん剤で発現する血小板減少を含む血液毒性が回避されており、有効性と安全性のバランスが改善され、長期に継続して治療することが可能となった。 化合物の組み合わせを改良した「モジュール創薬の代表例」。 手術後の微小がんの再発転移防止に最適で、高い延命効果が期待できる。 |
|
特許取得国(21年5月) |
日本、アメリカ、EU、中華人民共和国、オーストラリア、韓国、ロシア、中華民国、香港 |
(開発状況・今後の事業化)
米国にて固形がん(消化器がん)を対象に臨床第I相試験を進め、次試験の推奨用量と従来の5-FU系抗がん剤で発現していた血小板減少の副作用がないことを確認した。
現在、食事の影響試験が終了し、その総括作業と、治験責任医師との協議を行い、抗がん剤併用の第II相試験の治験計画を取りまとめ準備を進めている。
第I相試験と食事の影響試験の結果を中国臨床腫瘍学会(CSCO)と日本癌治療学会(JSCO)で発表した。
また、2020年5月には米国での臨床第I相試験結果の論文が米国のがん治療専門誌「Investigational New Drugs」に掲載され、下痢がない、白血球減少が少ない、血小板毒性が全くない、休薬期間が不要など安全性が確認され、高い延命効果が期待できるとの見解が発表された。こうした米国の臨床試験データに関心を寄せている中国の製薬会社との間で、米国と中国での共同開発余地についても協議中である。
現時点では「2022年3月期下期臨床第2相試験開始、2024年3月期下期臨床第3相試験開始、2027年3月期以降の申請及び上市」の予定である。
(特許関係)
DFP-11207は湿度に敏感で不安定なため、製剤技術の改良に注力した結果、安定製剤が完成したため、PCT(特許協力条約加盟国)出願と台湾出願を行った。2021年9月には、日本の特許庁より特許査定された。
今回の安定製剤の特許が成立すれば、更に長期に亘るDFP-11207の知財基盤が整うこととなる。
⑤「DFP-14927」
|
項目 |
概要 |
|
主な対象疾病 |
膵臓がん、胃がん、骨髄異形成症候群 |
|
既存薬の特徴など |
既存薬DFP-10917は投与には持続静注用ポーチを利用し、14日間連続の投与が必要で利便性の向上が必要であった。また対象疾患は血液がんのみであった。 |
|
モジュールの改良点・効果 |
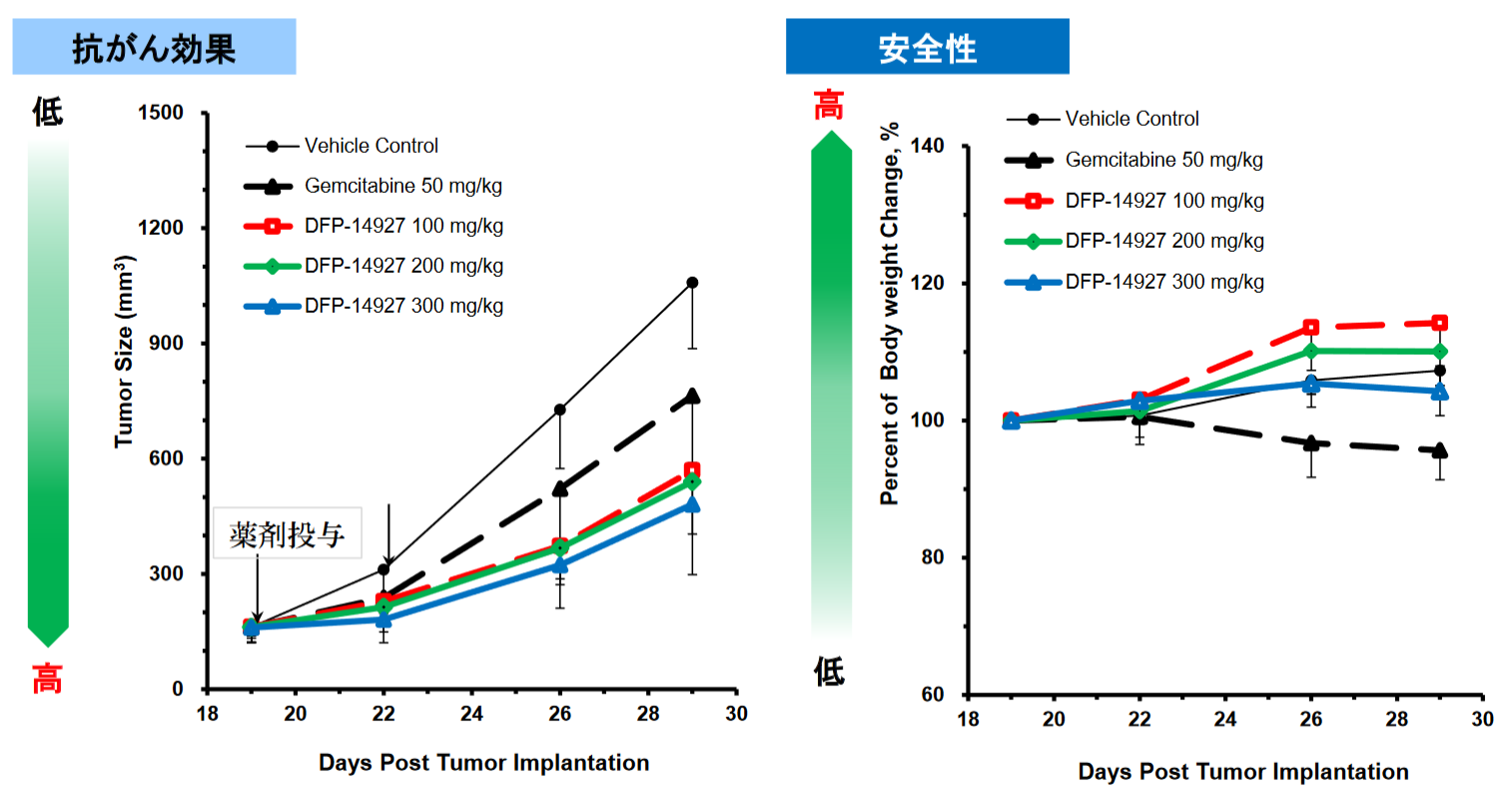
ポリエチレングリコール結合を行った抗がん剤候補物質DFP-14927は、DFP-10917の高分子デリバリーであり、がん組織へ選択的に集まり、がん細胞内で効果的にDFP-10917を放出することを可能とした。 また投与回数を週1回投与に減らし、投与経路も点滴静注とし、対象疾患は血液がんに加え、固形がんや骨髄異形成症候群に広がった。 加えて、膵臓がんの動物モデルでは、膵臓がんの標準化学療法剤であるゲムシタビンより効果、安全性が共に高いことが確認されている。 |
|
特許取得国(21年5月末) |
日本、アメリカ、中華人民共和国、オーストラリア、韓国、ロシア、香港 |
◎「DFP-14927」の実践動物での薬効の確認
膵臓がんの動物モデルにおいては、「DFP-14927」は膵臓がんの標準化学療法剤であるゲムシタビンよりも効果、安全性が共に高かった。
(同社資料より)
(開発状況・今後の事業化)
米国において前臨床試験が終了している。前臨床試験のデータでは、週1回投与で血液中濃度が長時間安定であることを確認しており、固形がんに対する抗腫瘍効果を認めている。
2018年3月に三洋化成工業(株)と共同開発契約を締結し、臨床第I相試験開始申請の準備を進めてきたが、2019 年1月18日、米国 FDAによる IND(Investigational New Drug:臨床試験用の新医薬品)の安全性審査が完了し、米国での臨床第I相試験の実施が許諾され、膵がん及び胃がんを含む消化器がん患者を対象に臨床第I相試験を開始した。
新型コロナウイルス感染拡大による影響によりコロナウイルス感染者数が多い地域において症例登録が鈍化しているが、現在の投与量付近で安全性が確認でき次第、最適のがん種を選定し、米国内の主要ながんセンターを複数追加のうえ、「2022年3月期下期に臨床第2相試験に相当する拡大試験へ移行」の予定である。
また、血液がんの骨髄異形成症候群(MDS)の臨床第1/2相試験の可能性を併せて検討する予定。
日本以外のテリトリーでの販売権に関しては、欧米の製薬会社及び中国の製薬会社 とライセンス契約交渉中である。
⑥「DFP-10825」
|
項目 |
概要 |
|
主な対象疾病 |
胃がん、卵巣がん、膵臓がんの腹膜播種転移 |
|
既存薬の特徴など |
基本薬siRNAは、基礎効果としては確実な阻害効果がみとめられるが、臨床効果としては全身投与での効果に難があった。 |
|
モジュールの改良点・効果 |
RNA干渉を利用した核酸医薬は、がん分子標的薬やがん免疫療法剤に次ぐ、次のがん治療薬として期待されている。核酸医薬DFP-10825は、がんの増殖に多大な影響を与える因子をRNA干渉で特異的に阻害させるために、全身投与ではなく腹腔内投与で効果を発揮できるように工夫している。卵巣がんや胃がん等の患者では、終末期になると胸水や腹水などの体液貯留(腹膜播種転移)が認められるが、腹腔内に直接注入して効果を発揮することにより、腹水をコントロールして苦しさを和らげ、延命につながることが期待される。 |
|
特許取得国(20年11月) |
日本、アメリカ、EU、中華人民共和国、オーストラリア、韓国、ロシア、中華民国、香港 |
(開発状況・今後の事業化)
すでに卵巣がん、胃がん及び膵がんで生じる腹水の原因となる腹膜播種転移に対する薬効試験と薬物動態試験を終え、原薬、DDS及び製剤などの治験薬の製造法についても現行の医薬品適正製造基準(cGMP)による予備的な検討を終えている。今後は、株式上場で得られた資金の一部を活用し、医薬品の安全性に関する非臨床試験の実施基準(GLP)による前臨床試験を追加した後、米国FDAにIND申請の上、米国で卵巣がん、胃がん及び膵がんの腹膜播種転移の患者を対象に臨床第I相試験を開始する予定である。出願中の各国の特許証も届いている。
原薬と治験製剤の準備を進めると共に、動物を用いた前臨床試験を進めており、「2022年3月期上期には臨床試験の準備に入る」予定である。
【1-5 バイオベンチャーとしての特徴・優位性】
バイオベンチャーである同社は以下のような特徴や優位性を有している。
➀モジュール創薬
前述のように、既存の薬剤等を「モジュール」(構成単位)で創意・工夫して組み立てることで特許化し、臨床上の有効性と安全性のバランスを向上させた新薬を生み出している。
➁抗がん剤開発への特化
未だに効果が限定的で多くの様々な副作用がある「抗がん剤」を対象にすることで、モジュール創薬による新薬開発を加速し、がん患者の社会生活の改善に貢献している。
③がん治療薬における優位性
同社では、標準療法後の「がん」の再発や難治性の患者にフォーカスした開発を進めている。したがって、標準治療薬投与後、効果が見いだせず治療薬が見つからない患者に向けた抗がん剤を提供する目的で開発を行っており、今後、開発品が上市された場合、標準療法後の治療薬として優位性が発生する。
④経験豊富なメンバーによる開発
長年にわたり抗がん剤の研究・開発に従事してきた製薬会社経験者と、がん患者のことを良く知る臨床医から構成されるメンバーで、確実に研究開発と資金確保を進め、アンメット・メディカル・ニーズに応えており、同社の強力な差別化要因、競争優位性となっている。
⑤外部資源の有効活用
工場や研究所を持たず、研究開発マネジメント業務に集中し、外部の受託機関などに委託して積極的な連携を図ることにより、効率的な運営を行っている。
2.業績動向
(1)2022年3月期第2四半期決算概要
①業績概況
|
|
21/3期2Q |
22/3期2Q |
前年同期比 |
|
事業収益 |
100 |
100 |
0 |
|
事業費用 |
563 |
654 |
+90 |
|
研究開発費 |
422 |
477 |
+54 |
|
その他販管費 |
140 |
177 |
+36 |
|
営業利益 |
-463 |
-554 |
-90 |
|
経常利益 |
-463 |
-555 |
-92 |
|
四半期純利益 |
-464 |
-557 |
-92 |
*単位:百万円
(事業収益)
日本ケミファとのライセンス契約によるマイルストーン収入を取得した。
(事業費用)
開発パイプラインの臨床試験における医療機関並びに症例数の増加、次試験に向けた治験薬となる原薬や製剤の製造などを進めた。
(営業利益)
営業損失は前年同期比90百万円拡大の5億54百万円となった。
②財務状態
◎主要BS
|
|
21年3月末 |
21年9月末 |
|
21年3月末 |
21年9月末 |
|
流動資産 |
2,115 |
1,696 |
負債合計 |
82 |
98 |
|
現預金 |
2,088 |
1,676 |
純資産合計 |
2,078 |
1,642 |
|
固定資産 |
45 |
44 |
利益剰余金 |
-4,484 |
-5,042 |
|
有形固定資産 |
41 |
40 |
負債純資産合計 |
2,161 |
1,741 |
|
資産合計 |
2,161 |
1,741 |
長短借入金残高 |
– |
– |
*単位:百万円
現預金の減少などで資産合計は前期末4億20百万円減少の17億41百万円。
利益剰余金が同5億57百万円減少した一方、新株予約権の行使により資本金・資本準備金が増加し、純資産は同4億35百万円減少し、16億42百万円。
自己資本比率は前期末比1.8ポイント低下し94.3%。
(2)2022年3月期業績見通し
|
|
21/3期 |
22/3期(予) |
前期比 |
|
事業収益 |
300 |
100 |
-200 |
|
事業費用 |
1,152 |
1,400 |
+247 |
|
研究開発費 |
866 |
1,090 |
+223 |
|
その他販管費 |
285 |
310 |
+24 |
|
営業損失 |
-852 |
-1,300 |
-447 |
|
経常損失 |
-859 |
-1,300 |
-440 |
|
当期純損失 |
-862 |
-1,300 |
-437 |
*単位:百万円
(事業収益)
ライセンス契約に伴うマイルストーン対価として前期比2億円減少の1億円を見込んでいる。
DFP-10917のマイルストーン対価による収益のほか、米国で臨床第III相試験を実施しているDFP-10917や、国内で臨床第II相試験を実施しているDFP-14323を含め、複数の抗がん剤候補化合物の臨床試験が進んでおり、新しいパートナーとの提携による契約一時金等の収益も期待している。今後、収益が確実になった段階で適時見通しを明らかにしていく予定。
(事業費用)
前期比2億50百万円増加の14億円の計画。
DFP-10917は米国における臨床第III相試験の症例登録をさらに進めると共に、DFP-14927の米国における臨床第I相試験を完了し、拡大試験に移行する予定。また、DFP-14323の国内における臨床第II相試験の症例登録の完了により、次の臨床第III相試験(大規模比較試験)は国内外の製薬企業と合同で取り組むことを含めて準備を進める。
日本ケミファ(株)と提携したDFP-17729は、国内における臨床第I/II相試験を進め、第II相試験部分の症例登録をする予定です。これらの開発パイプラインを着実に進めるため、研究開発費は増加する見込みである。
(営業損失)
前期比4億47百万円拡大の13億円の予想。
3.成長戦略
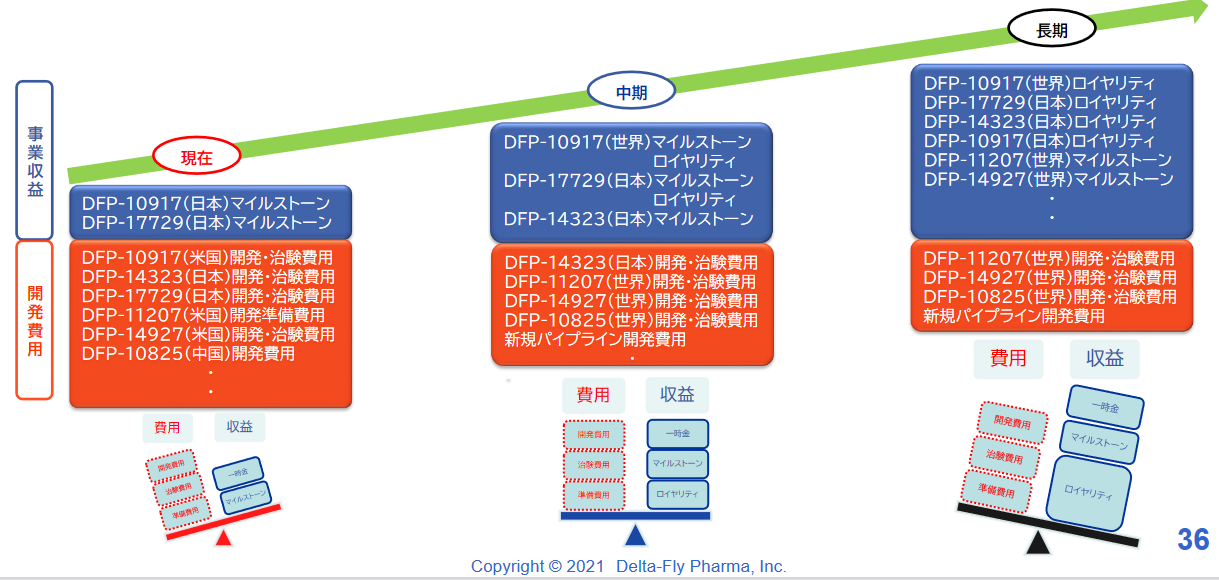
開発投資に関しては、企業価値向上を目指し、世界(欧米・アジア)・日本において手持資金、ライセンスフィー、資金調達等により開発投資を実施する。
現在は費用が先行し損失となっているが、計画的にパイプラインの上市を目指し、収支バランスを注視し、収益拡大を目指す。
(同社資料より)
臨床試験実施中の4品目および臨床試験準備中の2品目の開発を着実に進め、2022年度以降着実な上市を目指している。また収益の最大化を目指し、日本・中国・欧州・米国での提携パートナー確保にも注力する。
4.今後の注目点
引き続き、創業11年目に入った同社の具体的な成果として、最短での申請が予定されているDFP-10917の「2023年3月期上期の申請、同期下期の上市」に向けた着実な進捗を期待したい。
<参考:コーポレート・ガバナンスについて>
◎組織形態、取締役、監査役の構成
|
組織形態 |
監査役設置会社 |
|
取締役 |
8名、うち社外4名 |
|
監査役 |
3名、うち社外2名 |
◎コーポレート・ガバナンス報告書
最終更新日:2021年6月30日
<基本的な考え方>
当社は「「モジュール創薬」により、安心して家族のがん患者に勧められる治療法を提供する。」というミッションの下、株主をはじめ、顧客、取引先、従業員、地域社会等の全てのステークホルダーの利益を重視した経営を行うことが当社の使命であると考えています。そのためには、当社事業が安定的かつ永続的な発展を果たすことが不可欠であり、このような発展の基盤となる経営の健全性、透明性及び効率性が確保された体制の整備を進めることをコーポレート・ガバナンスの取組みに関する基本方針としています。
<コーポレート・ガバナンス・コードの各原則を実施しない理由>
「基本原則をすべて実施しています。」と記載している。